Cerveau en surcharge : glutamate, NMDA et brouillard mental dans le Covid long
La lumière devient agressive. Les sons ordinaires saturent. Une conversation de dix minutes peut déclencher un crash cognitif. Certains patients décrivent un cerveau « sans filtre », incapable de trier, de récupérer et de revenir au calme. Et si une partie du problème venait d'un cerveau bloqué dans un état d'hyperexcitabilité ? Le glutamate, les récepteurs NMDA, les astrocytes, la microglie, la voie kynurénine et les mitochondries offrent une grille de lecture plausible. Pas une explication unique. Encore moins une promesse de traitement.
Ce que vous allez comprendre
Il soutient l'attention, la mémoire et l'apprentissage. Il n'est pas toxique par nature. Il devient problématique quand sa régulation locale se dégrade.
Ces récepteurs aident le cerveau à apprendre. En activation prolongée, ils peuvent favoriser surcharge calcique, stress mitochondrial et stress oxydatif.
Microglie, cytokines, astrocytes, EAAT2 et voie kynurénine peuvent converger vers une excitabilité accrue chez certains profils.
Deux patients avec le même brouillard mental peuvent avoir des mécanismes dominants différents : perfusion, sommeil, dysautonomie, mastocytes, mitochondries ou neuroinflammation.
Aux personnes vivant avec brouillard mental, hypersensibilité sensorielle, fatigue mentale, douleur centrale ou crash cognitif dans un contexte de Covid long, d'EM/SFC, de fibromyalgie ou de maladie chronique complexe. Niveau intermédiaire : les mécanismes sont détaillés, mais chaque terme technique est défini.
📖 Glossaire bilingue
- Glutamate (glutamate) - principal neurotransmetteur excitateur du cerveau, indispensable à l'attention, à la mémoire et à l'apprentissage.
- Récepteur NMDA (N-methyl-D-aspartate receptor) - canal activé par le glutamate, perméable au calcium, impliqué dans la plasticité synaptique.
- Astrocyte (astrocyte) - cellule gliale qui nourrit les neurones, tamponne l'environnement synaptique et recapture le glutamate.
- Microglie (microglia) - cellule immunitaire résidente du cerveau, capable d'orchestrer une réponse inflammatoire locale.
- EAAT2/GLT-1 (excitatory amino acid transporter 2 / glutamate transporter 1) - transporteur astrocytaire majeur de recapture du glutamate.
- Voie kynurénine (kynurenine pathway) - voie de dégradation du tryptophane activée par l'inflammation, produisant des métabolites neuroactifs.
- Acide quinolinique (quinolinic acid) - métabolite de la voie kynurénine, agoniste des récepteurs NMDA dans certains contextes.
- Excitotoxicité (excitotoxicity) - modèle dans lequel une stimulation glutamatergique excessive endommage les neurones ou les synapses.
- Hypométabolisme (hypometabolism) - baisse de l'utilisation du glucose dans une région cérébrale, mesurable en TEP au 18F-FDG.
- PEM (post-exertional malaise) - aggravation retardée après effort physique, cognitif ou sensoriel, fréquente dans l'EM/SFC et certains Covid longs.
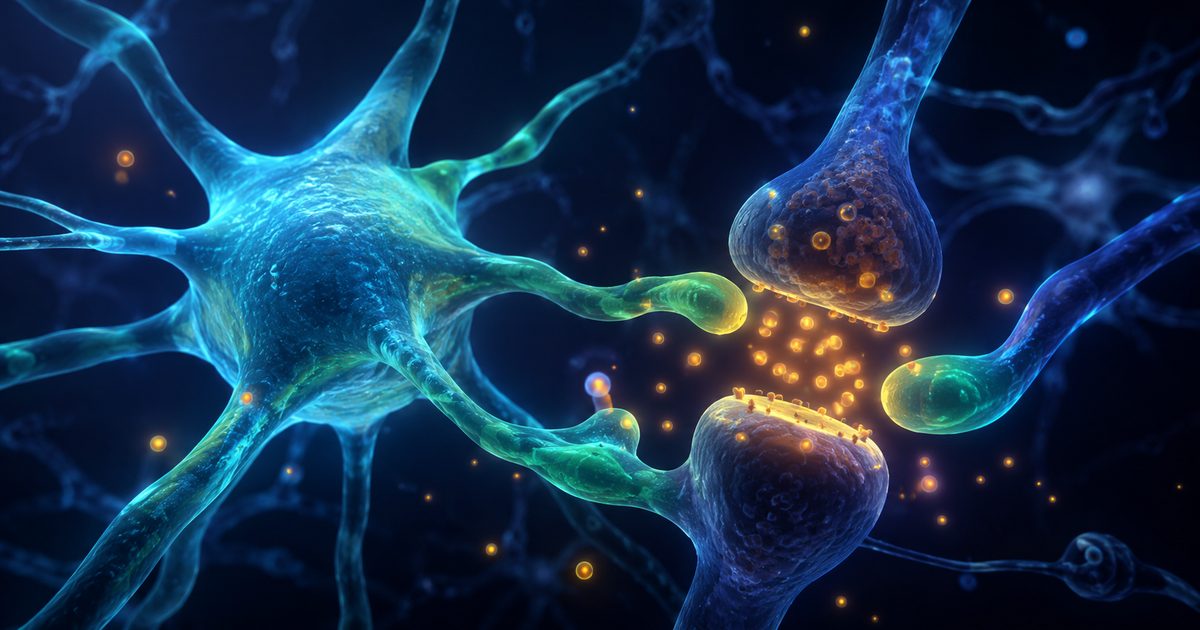
Le glutamate : le grand accélérateur du cerveau
🟢 Physiologie établie - neurosciencesLe glutamate est le principal signal excitateur du cerveau. Il permet d'allumer les réseaux de l'attention, de la mémoire et de l'apprentissage. Il n'est pas toxique par nature : le problème commence quand le signal dure trop longtemps ou n'est plus correctement recyclé.
Voir le détail ↓Le glutamate est le neurotransmetteur excitateur le plus abondant du système nerveux central. Il participe à la mémoire, à l'attention, à l'apprentissage et à la plasticité synaptique, c'est-à-dire la capacité des circuits cérébraux à se modifier avec l'expérience.
Le point critique est sa durée d'action. Le glutamate doit être libéré, transmettre l'information, puis disparaître rapidement de la fente synaptique. Trop peu de signal et le réseau devient lent. Trop de signal ou un signal trop prolongé et le cerveau peut entrer dans une logique d'hyperexcitabilité. Dans les modèles les plus sévères, cette stimulation excessive est appelée excitotoxicité[1].
Le maintien de cet équilibre repose sur un cycle en trois étapes :
① Le neurone pré-synaptique libère du glutamate dans la fente synaptique en réponse à un signal électrique. Le glutamate se fixe sur les récepteurs du neurone post-synaptique (AMPA, NMDA, kaïnate) et transmet l'information.
② Les astrocytes captent le glutamate excédentaire grâce à leurs transporteurs GLT-1/EAAT2, qui assurent une grande partie de la clairance synaptique. Le glutamate est converti en glutamine par la glutamine synthétase, puis remis à disposition des neurones.
③ La glutamine est renvoyée au neurone, qui la reconvertit en glutamate pour un nouveau cycle de neurotransmission. Ce circuit fermé - le cycle glutamate-glutamine - empêche l'accumulation de glutamate dans l'espace extracellulaire[5].
Les récepteurs NMDA : le portail calcique
🟢 Mécanisme établi - neurophysiologieLes récepteurs NMDA sont des portails à calcium. Ils s'ouvrent quand le glutamate se fixe et quand le frein magnésium se lève. C'est utile pour apprendre. En activation prolongée, ce même portail peut devenir une source de surcharge cellulaire.
Voir le détail ↓Le récepteur NMDA est un canal ionique particulier : il ne s'ouvre que lorsque deux conditions sont remplies simultanément. La première est la fixation du glutamate sur le récepteur. La seconde est une dépolarisation suffisante de la membrane pour déloger l'ion magnésium (Mg²⁺) qui bloque le canal au repos[7]. Ce double verrou fait du récepteur NMDA un détecteur de coïncidence, base moléculaire de la mémoire et de l'apprentissage.
En condition physiologique, ce mécanisme est finement régulé. Le calcium entre brièvement. La synapse se renforce ou s'ajuste. C'est l'un des socles de la mémoire et de la vigilance. Mais quand le glutamate persiste, ou quand le système inflammatoire modifie l'environnement synaptique, le verrou peut rester ouvert trop longtemps.
① Suractivation tonique. Le glutamate ambiant maintient une partie des récepteurs NMDA en activité prolongée, au-delà de la fenêtre physiologique de quelques millisecondes. Le canal reste ouvert plus longtemps, le calcium entre davantage[1].
② Surcharge calcique intraneuronale. Le calcium en excès active des cascades enzymatiques destructrices : calpaïnes (protéases), phospholipases (dégradation membranaire), endonucléases (fragmentation ADN). Les mitochondries, surchargées en calcium, dysfonctionnent et génèrent des espèces réactives de l'oxygène (ROS)[1].
③ Stress du réticulum endoplasmique et dommage synaptique. Selon l'intensité et la durée, la cellule peut subir une perte de synapses, une altération de la plasticité, un stress du réticulum endoplasmique et une baisse de performance énergétique. Dans un cerveau déjà vulnérable, cela peut se traduire par fatigue mentale, lenteur, hypersensibilité ou crash cognitif[1][8].
Le mécanisme du bloc magnésien voltage-dépendant du récepteur NMDA est un point clé de physiologie. Un déficit magnésien peut théoriquement affaiblir ce frein naturel, mais l'inférence clinique doit rester prudente[7]. Le magnésium sérique reflète mal les compartiments intracellulaires, le statut magnésien est complexe, et un bloc NMDA physiologique ne démontre pas un état excitotoxique clinique. Corriger une carence documentée est rationnel ; cela ne montre pas qu'une supplémentation améliore le brouillard mental du Covid long.
Comment l'inflammation peut pousser le système vers l'hyperexcitabilité
🟠 Modèle intégratif - données précliniques et observationnellesL'inflammation peut modifier la façon dont le cerveau gère le glutamate. Microglie activée, cytokines, astrocytes réactifs, transporteurs EAAT2 freinés et voie kynurénine peuvent pousser certains réseaux vers une hyperexcitabilité durable.
Voir le détail ↓Dans ce modèle, le problème ne vient pas d'une seule molécule. Il vient d'un réseau. La microglie peut rester activée après un signal infectieux. Les cytokines modifient l'environnement synaptique. Les astrocytes deviennent moins efficaces pour tamponner le glutamate. Les mitochondries produisent moins d'énergie et plus de ROS. Chaque étage peut amplifier le suivant.
Le transporteur GLT-1, aussi appelé EAAT2, reste une pièce centrale. Exprimé surtout sur les prolongements astrocytaires qui entourent les synapses, il assure une grande partie de la recapture du glutamate. Quand cette recapture ralentit, le glutamate extracellulaire peut durer plus longtemps, donc stimuler plus fortement les récepteurs NMDA[5].
Plusieurs mécanismes convergent pour inhiber GLT-1 en contexte de neuroinflammation post-infectieuse :
① Les cytokines pro-inflammatoires (TNF-α, IL-1β, IL-6) réduisent l'expression transcriptionnelle de GLT-1 et accélèrent son internalisation depuis la membrane astrocytaire. Moins de transporteurs en surface signifie moins de glutamate recyclé[5].
② Le stress oxydatif peut altérer la fonction des transporteurs du glutamate dans plusieurs modèles biologiques. Les espèces réactives de l'oxygène (ROS) générées par la neuroinflammation chronique peuvent donc créer une boucle de surcharge excitatrice : moins de recapture → glutamate plus durable → activité NMDA accrue → stress cellulaire → encore moins de recapture[5].
③ Le SARS-CoV-2 peut perturber des cellules du système nerveux central. Proust et al. (2023) ont étudié l'effet de plusieurs variants sur des modèles cellulaires incluant astrocytes, péricytes, cellules endothéliales et microglie, avec mesure de la concentration extracellulaire de glutamate. Cette donnée soutient la plausibilité d'un effet sur l'environnement neuroglial ; elle ne démontre pas à elle seule une défaillance persistante de recapture du glutamate chez les patients en Covid long[2].
④ La voie kynurénine ajoute un second levier. L'inflammation peut détourner le tryptophane vers la voie kynurénine. Certains métabolites sont neuroactifs. L'acide quinolinique, notamment, agit comme agoniste NMDA et peut soutenir une logique excitotoxique dans plusieurs modèles neuroinflammatoires[18][19]. Dans le Covid long, les données sur cette voie existent, mais restent hétérogènes : elles soutiennent une hypothèse plausible, pas un biomarqueur clinique robuste.
⑤ Un modèle inflammatoire viral renforce la cohérence. Shi et al. (2025) ont montré dans une co-culture neurone-microglie-astrocyte que Poly I:C, un analogue viral, déclenche une activation gliale, augmente TNF-α et IL-6, diminue GLT-1 et perturbe l'intégrité synaptique[6]. Ce modèle n'est pas le Covid long humain. Il montre surtout qu'un signal viral peut déplacer l'équilibre glial-glutamatergique.
Boucle simplifiée : inflammation → microglie activée → cytokines → astrocytes moins efficaces → recapture EAAT2 réduite → glutamate plus durable → NMDA plus actif → calcium → mitochondries sous stress → ROS → nouvelle inflammation.
Voie parallèle : inflammation → voie kynurénine → acide quinolinique → activation NMDA → bruit neuronal et stress cellulaire.
Fait établi : astrocytes, microglie et mitochondries dialoguent en permanence avec les synapses. Hypothèse étayée : certains profils post-infectieux pourraient rester bloqués dans une boucle neuroinflammatoire glutamatergique. Point débattu : la fréquence réelle de ce profil dans le Covid long et sa mesure fiable en clinique.
À ce stade, le modèle décrit surtout une convergence mécanistique théorique compatible avec certains profils cliniques, plutôt qu'un mécanisme démontré chez l'humain vivant atteint de Covid long.
Le brouillard mental fluctue d'un jour à l'autre. Un suivi de la clarté mentale, de l'énergie et du sommeil permet de repérer les patterns et d'objectiver les améliorations ou les dégradations lors de l'introduction d'un traitement.
Commencer le suivi avec BoussoleLe cerveau « sans filtre »
🟠 Lecture clinique - mécanisme plausible, profils hétérogènesUn cerveau hyperexcitable ne se contente pas d'être fatigué. Il filtre moins bien. Il hiérarchise moins bien. Il transforme parfois des signaux ordinaires en charge excessive : lumière, bruit, douleur, mouvement, conversation, lecture, écran, décision.
Cette grille de lecture aide à relier plusieurs symptômes souvent décrits séparément :
① Hypersensibilité sensorielle. La lumière paraît trop forte, les sons trop proches, les environnements complexes trop coûteux. Le problème peut venir d'une baisse du filtrage sensoriel, pas seulement d'une fragilité psychologique.
② Hypervigilance et excitation interne. Certains patients se sentent épuisés mais « branchés », incapables de redescendre. Cela peut ressembler à de l'anxiété. Cela peut aussi impliquer une physiologie réelle : noradrénaline, microglie, sommeil fragmenté, douleur centrale et circuits glutamatergiques.
③ Douleur centrale. Dans la sensibilisation centrale, le système nerveux amplifie la douleur même quand le signal périphérique initial ne suffit plus à expliquer l'intensité ressentie. Les récepteurs NMDA participent à cette plasticité douloureuse dans plusieurs modèles de douleur chronique[10].
④ Crash cognitif. L'effort mental consomme de l'énergie, augmente l'activité synaptique et peut aggraver une boucle déjà instable. Chez certains profils, lire, travailler ou tenir une réunion peut produire une aggravation retardée, proche du malaise post-effort décrit dans l'EM/SFC.
Les deux peuvent coexister. Mais réduire l'hypervigilance, l'intolérance au bruit ou le crash cognitif à une cause psychologique unique est un raccourci. Un système nerveux inflammatoire, mal reposé, dysautonomique et énergétiquement fragile peut produire des sensations très proches de l'anxiété sans que l'anxiété soit le mécanisme principal.
Le cercle vicieux : surcharge excitatrice, stress oxydatif et transporteurs
🟠 Mécanismes documentés - intégration théoriqueUne surcharge excitatrice peut former une boucle auto-entretenue. Le glutamate durable favorise le stress oxydatif, qui peut altérer les transporteurs chargés de le recycler, ce qui aggrave encore l'accumulation. Cette boucle est un modèle plausible de persistance, pas l'explication unique du brouillard mental.
Voir le détail ↓La persistance du brouillard mental post-COVID pourrait, chez certains profils, être entretenue par une boucle entre excitation synaptique prolongée, stress oxydatif et transporteurs astrocytaires moins efficaces. Ce modèle explique une partie des données mécanistiques, mais il reste à valider chez des cohortes cliniques stratifiées. Le terme excitotoxicité doit être réservé aux modèles sévères de dommage cellulaire, pas utilisé comme synonyme automatique de brouillard mental.
Le mécanisme en boucle :
① Glutamate accumulé → suractivation NMDA → influx calcium : la cascade décrite dans la section précédente.
② Calcium en excès → dysfonction mitochondriale → production de ROS : les mitochondries surchargées en calcium produisent des espèces réactives de l'oxygène au lieu d'ATP[1].
③ ROS → oxydation des transporteurs GLT-1 → moins de recapture du glutamate : les résidus cystéine critiques de GLT-1 sont oxydés, la protéine perd sa fonction de transport[5].
④ Moins de recapture → plus de glutamate dans la fente → retour à l'étape ①
Pimenta et al. (2023) soutiennent ce modèle chez l'animal : l'inflammation induite par le SARS-CoV-2 s'accompagne d'une augmentation du glutamate extracellulaire, de marqueurs de stress oxydatif et d'une réduction de l'expression de GLT-1. La transposition au brouillard mental humain reste une hypothèse étayée, pas une validation clinique[3].
Pourquoi des molécules très différentes convergent vers cet axe
🟠 Pharmacologie mécanistique - pas une recommandation de traitementCe tableau explique pourquoi des molécules très différentes croisent parfois la boucle glutamate/NMDA. Il ne conclut pas à une efficacité thérapeutique. Les niveaux de données sont hétérogènes, les indications réglementaires diffèrent, et plusieurs usages seraient hors AMM dans le Covid long.
Une convergence mécanistique ne prédit ni efficacité clinique, ni balance bénéfice-risque favorable. Certaines molécules peuvent même aggraver fatigue, dysautonomie, cognition, sommeil ou hypersensibilité chez certains profils.
La question clinique n'est donc pas « quelle molécule bloque NMDA ? ». La vraie question est : quel mécanisme domine chez ce patient, à ce moment, avec quels risques ?
Le piège serait de croire que toutes ces molécules font la même chose. Elles convergent parfois vers l'axe glutamate/NMDA, mais elles n'agissent pas au même endroit de la boucle. Certaines modulent directement le récepteur NMDA. D'autres réduisent l'entrée excitatrice en amont. D'autres ciblent le stress oxydatif, la noradrénaline préfrontale ou l'état de vigilance.
| Molécule | Mécanisme principal | Lien avec l'axe glutamate/NMDA | Nuances / limites |
|---|---|---|---|
| Kétamine | Antagonisme NMDA non compétitif, effets rapides sur certains circuits glutamatergiques et la plasticité synaptique. | Agit au niveau du récepteur NMDA, avec effets secondaires dissociatifs et cardiovasculaires possibles. | Indications précises selon pays et contexte. Ce n'est pas un traitement validé du brouillard mental du Covid long. Risque élevé de mésusage et de simplification médiatique[20]. |
| Mémantine | Antagoniste NMDA de faible affinité, non compétitif, utilisé dans la maladie d'Alzheimer. | Peut réduire une activation NMDA excessive tout en préservant une partie de la transmission physiologique, selon son modèle pharmacologique[17]. | Signal indirect dans la fibromyalgie surtout sur douleur/impact global, pas validation cognitive Covid long[9]. Prudence rénale et interactions. |
| Dextrométhorphane | Antitussif avec effets NMDA, sigma-1 et monoaminergiques selon dose et formulation. | Peut moduler les courants NMDA toniques et certains circuits glutamatergiques[21]. | Risque d'interactions sérotoninergiques, mésusage, variabilité métabolique CYP2D6. Pas de conclusion clinique Covid long. |
| Magnésium | Cofacteur minéral, bloc physiologique voltage-dépendant du canal NMDA au repos. | Participe au frein naturel de l'entrée calcique via NMDA[7]. | Corriger une carence documentée est rationnel. Supplémenter sans indication ne démontre pas une amélioration du brouillard mental. Prudence en insuffisance rénale. |
| Prégabaline | Ligand α2δ des canaux calciques voltage-dépendants, utilisé dans certaines douleurs neuropathiques. | Réduit la libération de neurotransmetteurs excitateurs en amont et peut influencer le couplage α2δ-1/NMDA dans des modèles de douleur[10][11][12][13]. | Somnolence, confusion, dépendance, sevrage, adaptation rénale. En France, conditions de prescription renforcées[16]. |
| NAC | Précurseur du glutathion, modulation redox, interaction avec le système cystine/glutamate. | Peut influencer l'homéostasie glutamate/glutathion et le stress oxydatif, donc agir plutôt en aval ou en parallèle de la boucle NMDA[22]. | Données Covid long surtout ouvertes ou indirectes. Les effets attendus dépendent du profil redox, inflammatoire et digestif. |
| Guanfacine | Agoniste α2A-adrénergique, renforcement des réseaux préfrontaux et réduction de certains signaux de stress neuronal. | Ne bloque pas NMDA directement. Peut stabiliser le cortex préfrontal et réduire le bruit noradrénergique qui aggrave la cognition en contexte inflammatoire[23]. | Petite série ouverte guanfacine + NAC : 12 patients, absence d'essai randomisé, forte possibilité d'effet placebo, de sélection ou de régression vers la moyenne[24]. Résultats non reproductibles à ce stade. Risque d'hypotension, sédation, interaction avec dysautonomie. |
| Clonidine | Agoniste α2-adrénergique plus global, baisse du tonus sympathique. | Agit sur l'hyperadrénergie et l'état d'alerte plus que sur NMDA. Peut réduire un étage amont de la surcharge chez certains profils dysautonomiques. | Hypotension, bradycardie, sédation, rebond hypertensif si arrêt brutal. Pas de validation spécifique Covid long neurocognitif. |
Fait établi : ces molécules ont des cibles pharmacologiques différentes. Hypothèse étayée : elles peuvent influencer différentes zones d'une même boucle excitabilité-inflammation-énergie. Point débattu : savoir si cette convergence se traduit par un bénéfice clinique reproductible dans le Covid long.
Le piège : même symptôme ne veut pas dire même mécanisme
🟠 Point clinique central - hétérogénéité majeureLe brouillard mental n'est pas un mécanisme. C'est une plainte clinique. Elle peut venir de plusieurs routes biologiques différentes, parfois superposées chez la même personne.
Deux patients peuvent dire exactement la même phrase - « je n'arrive plus à penser » - et ne pas avoir le même problème dominant. Chez l'un, le facteur principal peut être une hypoperfusion cérébrale liée à la dysautonomie. Chez l'autre, une hyperexcitabilité glutamatergique. Chez un troisième, un sommeil fragmenté, une activation mastocytaire, une inflammation microgliale, une fatigue mitochondriale ou une hyperadrénergie.
Patient A : lever debout → tachycardie → baisse de perfusion cérébrale → brouillard mental.
Patient B : bruit + lumière → surcharge sensorielle → NMDA/glutamate → crash cognitif.
Patient C : sommeil fragmenté → microglie plus réactive → douleur centrale + fatigue mentale.
Patient D : mastocytes/histamine → hypervigilance + céphalées + intolérance sensorielle.
C'est ici que la pharmacologie devient piégeuse. Une réponse positive à une molécule ne valide pas un mécanisme universel. Si une personne se sent mieux avec une molécule qui touche indirectement l'axe NMDA, cela peut signifier que cette boucle comptait chez elle. Cela ne signifie pas que tous les brouillards mentaux sont NMDA-dépendants, ni que la même molécule conviendra aux autres.
La fibromyalgie, l'EM/SFC et le Covid long illustrent ce problème. Ces tableaux peuvent partager douleur centrale, hypersensibilité, fatigue mentale et troubles du sommeil. Ils peuvent aussi diverger par leurs déclencheurs, leur dysautonomie, leur immunologie, leur perfusion cérébrale ou leur terrain hormonal. Le bon modèle est donc un modèle intégratif, pas une étiquette unique.
Raisonner par sous-profils : profil dysautonomique, profil neuroinflammatoire, profil sommeil, profil mastocytaire, profil douleur centrale, profil mitochondrial, profil hyperadrénergique. Ces profils peuvent se chevaucher. L'objectif n'est pas de cocher une case, mais de repérer la boucle dominante et les facteurs aggravants.
Ce que la science montre... et ce qu'elle ne montre pas
🟠 Synthèse critique - données solides, extrapolations et limitesLa partie solide est la physiologie. Le glutamate est le principal neurotransmetteur excitateur. Les récepteurs NMDA laissent entrer du calcium. Les astrocytes participent à la recapture du glutamate. La microglie et les cytokines peuvent modifier la transmission synaptique. Le stress mitochondrial et les ROS peuvent amplifier l'inflammation. Ces éléments ne sont pas controversés isolément[5][7].
La partie plausible concerne leur combinaison dans le Covid long. Des travaux associent troubles cognitifs post-COVID, perturbation de la barrière hémato-encéphalique et signaux compatibles avec une neurotoxicité glutamatergique dans certains contextes[1]. Des modèles animaux ou cellulaires suggèrent que l'infection ou des signaux viraux peuvent activer la glie, modifier le glutamate et réduire GLT-1/EAAT2[2][3][6].
L'imagerie apporte un signal fonctionnel, pas une cause unique. Des études en TEP au 18F-FDG ont rapporté des profils d'hypométabolisme cérébral chez certains patients Covid long, notamment dans des régions olfactives, limbico-paralimbiques, cérébelleuses ou du tronc cérébral[4][25]. Mais un hypométabolisme ne dit pas automatiquement « glutamate ». Il peut refléter perfusion, inflammation, activité neuronale, énergie mitochondriale ou interactions entre ces niveaux.
La voie kynurénine est cohérente, mais pas simple. Certaines équipes proposent qu'une activation inflammatoire de cette voie puisse produire des métabolites neuroactifs, dont l'acide quinolinique, susceptible d'activer NMDA[18]. Mais les analyses du Covid long sont hétérogènes et ne permettent pas aujourd'hui d'utiliser cette voie comme test clinique robuste[19].
La spectroscopie IRM et l'imagerie neuroinflammatoire ouvrent une piste intéressante. Une étude préliminaire a mesuré des variations de neurochimiques cérébraux dans l'EM/SFC et le Covid long, avec des signaux glutamatergiques dans certaines régions[14]. Des données TEP antérieures dans l'EM/SFC ont aussi rapporté une activation microgliale/astrocytaire corrélée à certains symptômes cognitifs[15]. C'est important, mais les effectifs restent limités. Ce n'est pas encore un biomarqueur clinique de routine.
La partie la plus fragile est thérapeutique. Comprendre une boucle ne suffit pas à savoir comment l'interrompre. Les essais sont rares, les sous-groupes mal définis, les critères de jugement variables et les risques médicamenteux réels. Une molécule peut avoir un mécanisme séduisant et un effet clinique nul, ou un effet chez un sous-groupe non identifié.
Données solides : physiologie glutamate/NMDA, rôle des astrocytes, lien inflammation-neurotransmission, existence de symptômes cognitifs et sensoriels dans le Covid long. Hypothèses plausibles : boucle microglie-astrocytes-glutamate-NMDA-mitochondries chez certains profils. Extrapolations : appliquer directement des données douleur, dépression, Alzheimer ou modèles animaux au Covid long. Limite majeure : absence de biomarqueur clinique robuste permettant d'identifier facilement un sous-type glutamatergique.
🟢 Établi : glutamate, récepteurs NMDA, rôle des astrocytes, activation microgliale et interaction inflammation-neurotransmission.
🟡 Plausible : boucle persistante post-infectieuse, recapture EAAT2 diminuée, voie kynurénine produisant des métabolites excitateurs.
🟠 Exploratoire : sous-type glutamatergique du Covid long, ciblage pharmacologique personnalisé, usage de marqueurs d'imagerie ou de neurochimie pour stratifier les patients.
🔴 Non démontré : biomarqueur clinique fiable, traitement NMDA validé du Covid long, protocole guanfacine/NAC ou mémantine généralisable.
① Synapse en surcharge : glutamate persistant, NMDA ouvert, calcium entrant. ② Astrocyte protecteur puis débordé : EAAT2 actif puis réduit par cytokines/ROS. ③ Boucle microglie-mitochondries : inflammation, ROS, recapture diminuée. ④ Cerveau sans filtre : bruit sensoriel entrant, cortex préfrontal saturé, récupération retardée.
Fait établi : le glutamate est un neurotransmetteur excitateur majeur. Les récepteurs NMDA contrôlent une entrée de calcium impliquée dans la plasticité synaptique. Les astrocytes recapturent le glutamate via EAAT2/GLT-1. La microglie, les cytokines, le sommeil, les mitochondries et la perfusion cérébrale modifient le fonctionnement cognitif.
Hypothèse étayée : chez certains profils Covid long, EM/SFC ou fibromyalgie, une boucle neuroinflammation → astrocytes → glutamate → NMDA → calcium → mitochondries → ROS pourrait contribuer au brouillard mental, à l'hypersensibilité sensorielle, à la douleur centrale et au crash cognitif. Cette hypothèse reste une convergence mécanistique compatible avec certains profils, pas un mécanisme clinique démontré chez tous les patients.
Spéculation à surveiller : l'idée qu'un patient donné serait « glutamate/NMDA » sans mesure robuste, ou qu'une réponse à une molécule suffirait à identifier le mécanisme. Le risque clinique est de transformer une grille de lecture en raccourci thérapeutique.
Ce qu'il faut retenir
Le glutamate et les récepteurs NMDA offrent peut-être une grille de lecture utile pour comprendre certains symptômes neurocognitifs du Covid long. Brouillard mental, hypersensibilité sensorielle, hypervigilance, douleur centrale, fatigue mentale et crash cognitif peuvent parfois s'inscrire dans un modèle d'hyperexcitabilité neuroinflammatoire.
Mais ce modèle n'est probablement qu'un réseau biologique parmi d'autres. Microglie, astrocytes, mitochondries, dysautonomie, perfusion cérébrale, sommeil, inflammation, mastocytes et métabolisme interagissent ensemble. Réduire le symptôme à NMDA serait aussi fragile que le réduire à l'anxiété.
Comprendre un symptôme ne signifie pas encore savoir quelle molécule utiliser.Questions fréquentes
Pourquoi certains patients décrivent-ils un cerveau « sans filtre » ?
Le brouillard mental post-COVID est-il toujours lié au glutamate ?
La mémantine peut-elle améliorer le brouillard mental du Covid long ?
Pourquoi parler de guanfacine, NAC ou clonidine dans un article sur NMDA ?
Une réponse positive à une molécule valide-t-elle le mécanisme NMDA ?
Existe-t-il un biomarqueur clinique fiable du profil glutamate/NMDA ?
Suivre la clarté mentale au quotidien aide à objectiver les fluctuations sans conclure trop vite à une cause unique. Repérez les facteurs d'amélioration et préparez des consultations plus structurées.
Commencer avec BoussoleSources
- Chaganti J, Schroder L, Engstrom C, et al. Blood-brain barrier disruption and glutamatergic excitotoxicity in post-acute sequelae of SARS-CoV-2 infection cognitive impairment. Front Neurol. 2024;15:1350848. Chaganti et al., 2024 - PubMed PMID 38756214
- Proust A, Barat C, Leblanc É, Bhagwat A, Bhargava R, et al. Differential effects of SARS-CoV-2 variants on central nervous system cells and blood-brain barrier functions. J Neuroinflammation. 2023;20(1):183. Proust et al., 2023 - PubMed PMID 37537664
- Pimenta MAS, Barbosa EJR, Elias-Oliveira J, et al. SARS-CoV-2 infection triggers neuroinflammation and glutamate excitotoxicity in animal model. Inflamm Res. 2023;72(10-11):1847-1859. Pimenta et al., 2023 - PubMed PMID 37837557
- Hugon J, Msika EF, Queneau M, Farid K, Paquet C. Cognitive decline and brainstem hypometabolism in long COVID: A case series. Brain Behav. 2022;12(4):e2513. Hugon et al., 2022 - PubMed PMID 35290729
- Dahlmanns M, Dahlmanns JK, Schmidt CC, et al. Glutamate transporter-mediated glutamate uptake and its implications for neurodegenerative diseases. Front Biosci (Landmark Ed). 2023;28(3):57. Dahlmanns et al., 2023 - PubMed PMID 37005761
- Shi J, Chen Y, et al. Ceftriaxone attenuates Poly I:C-induced neuroinflammation in vitro by modulating glutamate transport, synaptic integrity, and immunometabolic reprogramming. Front Cell Neurosci. 2025;19:1684398. Shi et al., 2025 - Frontiers
- Kirkland AE, Sarlo GL, Holton KF. The role of magnesium in neurological disorders. Nutrients. 2018;10(6):730. Kirkland et al., 2018 - PubMed PMID 29882776
- Prentice H, Modi JP, Wu JY. Mechanisms of neuronal protection against excitotoxicity, endoplasmic reticulum stress, and mitochondrial dysfunction in stroke and neurodegenerative diseases. Oxid Med Cell Longev. 2015;2015:964518. Voir aussi : Prentice et al. Taurine protects against glutamate excitotoxicity. In: Taurine 10. Adv Exp Med Biol. 2017;975:597-606. Prentice et al., 2017 - PubMed PMID 28849455
- Olivan-Blázquez B, Herrera-Mercadal P, Puebla-Guedea M, et al. Efficacy of memantine in the treatment of fibromyalgia: A double-blind, randomised, controlled trial with 6-month follow-up. Pain. 2014;155(12):2517-2525. Olivan-Blázquez et al., 2014 - PubMed PMID 25218600
- Deng M, Chen SR, Pan HL. Presynaptic NMDA receptors control nociceptive transmission at the spinal cord level in neuropathic pain. Cell Mol Life Sci. 2019;76(10):1889-1899. Deng et al., 2019 - PubMed PMID 30788514
- Chen Y, Chen SR, Chen H, Zhang J, Pan HL. Increased α2δ-1-NMDA receptor coupling potentiates glutamatergic input to spinal dorsal horn neurons in chemotherapy-induced neuropathic pain. J Neurochem. 2019;148(2):252-263. Chen et al., 2019 - PubMed PMID 30431158
- Chincholkar M. Analgesic mechanisms of gabapentinoids and effects in experimental pain models: a narrative review. Br J Anaesth. 2018;120(6):1315-1334. Chincholkar, 2018 - PubMed PMID 29793598
- Suto T, Eisenach JC, Hayashida K. Gabapentin increases extracellular glutamatergic level in the locus coeruleus via astroglial glutamate transporter-dependent mechanisms. Neuropharmacology. 2014;81:95-100. Suto et al., 2014 - PubMed PMID 24495399
- Thapaliya K, Marshall-Gradisnik S, Barth M, et al. Imbalanced Brain Neurochemicals in Long COVID and ME/CFS: A Preliminary Study Using MRI. Am J Med. 2024;138(3):567-574. DOI: 10.1016/j.amjmed.2024.04.007. Thapaliya et al., 2024 - PubMed PMID 38588934
- Nakatomi Y, Mizuno K, Ishii A, et al. Neuroinflammation in Patients with Chronic Fatigue Syndrome/Myalgic Encephalomyelitis: An ¹¹C-(R)-PK11195 PET Study. J Nucl Med. 2014;55(6):945-950. DOI: 10.2967/jnumed.113.131045. Nakatomi et al., 2014 - PubMed PMID 24665088
- ANSM. Prégabaline : nouvelles conditions de prescription et délivrance pour limiter le mésusage, l'abus et la dépendance. Information transmise sous l'autorité de l'ANSM, 2021. ANSM - prégabaline
- Base de Données Publique des Médicaments. Mémantine : résumé des caractéristiques du produit. BDPM - mémantine
- Liu Y, et al. Tryptophan Metabolism in Central Nervous System Diseases: Pathophysiology and Potential Therapeutic Strategies. Front Immunol. 2023. PubMed PMID 37191427
- Wang B, et al. The kynurenine pathway in COVID-19 neuropathogenesis. Int J Mol Sci. 2024. PMC - voie kynurénine et COVID-19
- Aleksandrova LR, Phillips AG, Wang YT. The mechanisms behind rapid antidepressant effects of ketamine: a systematic review with a focus on molecular neuroplasticity. Front Neuroanat. 2022. PMC - kétamine et neuroplasticité
- Seshadri A, et al. Modulating tonic NMDA receptor currents: mechanistic insights into ketamine, esketamine, and dextromethorphan. Neuropsychopharmacology. 2026. PubMed PMID 41577431
- Lewerenz J, Maher P. System x(c)-: cystine/glutamate antiporter and CNS redox/glutamate homeostasis. Antioxid Redox Signal. 2011. PubMed PMID 21564084
- Arnsten AFT, Datta D, Del Tredici K, Braak H. Scientific rationale for the use of α2A-adrenoceptor agonists in treating neuroinflammatory cognitive disorders. Mol Psychiatry. 2023. PubMed PMID 37029295
- Fesharaki-Zadeh A, Lowe N, Arnsten AFT. Clinical experience with the α2A-adrenoceptor agonist guanfacine and N-acetylcysteine for cognitive deficits in Long-COVID19. Neuroimmunology Reports. 2023;3:100154. PMC - guanfacine + NAC
- Guedj E, Campion JY, Dudouet P, et al. 18F-FDG brain PET hypometabolism in patients with long COVID. Eur J Nucl Med Mol Imaging. 2021;48:2823-2833. PubMed PMID 33501506