Gabapentinoïdes et douleur chronique : comprendre la neuromodulation
La prégabaline (Lyrica®) et la gabapentine (Neurontin®) comptent parmi les médicaments les plus prescrits dans la douleur chronique : fibromyalgie, douleur neuropathique, séquelles de zona. Pourtant, leur mécanisme d'action reste souvent mal compris, même par les prescripteurs. Ces deux molécules ciblent une protéine précise — la sous-unité α2δ des canaux calciques — au carrefour des circuits de la douleur. Comprendre comment la douleur est modulée par le système nerveux permet de saisir pourquoi ces molécules fonctionnent, et pourquoi elles ne sont pas des antalgiques classiques.
Ce que vous allez comprendre
C'est un dysfonctionnement mesurable des circuits de modulation de la douleur dans le système nerveux central.
Les gabapentinoïdes se lient à cette protéine sur les canaux calciques et réduisent la libération de neurotransmetteurs excitateurs dans la moelle épinière.
Même cible, mais biodisponibilité, pharmacocinétique et puissance de liaison très différentes.
Réduction modeste de la douleur dans les essais. Risque de mésusage et de dépendance, surtout à hautes doses.
Vous prenez de la prégabaline ou de la gabapentine (ou on vous en a proposé) et vous souhaitez comprendre comment ces molécules agissent réellement sur votre douleur chronique, au-delà du simple « ça calme les nerfs ».
📖 Glossaire bilingue : termes clés de cet article
- Gabapentinoïdes (gabapentinoids) : classe pharmacologique regroupant la prégabaline et la gabapentine
- Sous-unité α2δ (α2δ subunit) : protéine auxiliaire des canaux calciques voltage-dépendants, cible moléculaire des gabapentinoïdes
- Nociception (nociception) : détection et transmission des stimuli potentiellement nocifs par le système nerveux
- Corne dorsale (dorsal horn) : zone postérieure de la moelle épinière où les fibres de la douleur font synapse
- Modulation descendante (descending modulation) : circuits cérébraux (PAG → RVM → moelle) qui amplifient ou freinent la douleur
- Sensitisation centrale (central sensitization) : augmentation pathologique de la réactivité des neurones de la moelle épinière à la douleur
- Douleur nociplastique (nociplastic pain) : troisième catégorie de douleur, liée à un traitement central altéré sans lésion identifiable
- Wind-up : amplification progressive de la réponse d'un neurone spinal à une stimulation répétée
- PAG (periaqueductal gray) : substance grise périaqueducale, centre de contrôle de la modulation descendante
- RVM (rostral ventromedial medulla) : relais bulbaire contenant les cellules ON (pro-douleur) et OFF (anti-douleur)

Comment naît la douleur : de la périphérie à la moelle
Avant de comprendre comment les gabapentinoïdes agissent, il faut comprendre le circuit qu'ils ciblent. La douleur commence dans les tissus périphériques : une brûlure, une pression excessive, une inflammation activent des récepteurs spécialisés appelés nocicepteurs, situés aux terminaisons des fibres nerveuses C (lentes, douleur sourde) et Aδ (rapides, douleur aiguë).
Ces fibres transmettent le signal jusqu'à la corne dorsale de la moelle épinière, une zone de tri et de relais. C'est ici que le signal douloureux fait sa première synapse : la fibre périphérique libère des neurotransmetteurs excitateurs (glutamate, substance P, CGRP) qui activent le neurone de deuxième ordre. Ce neurone projette ensuite vers le thalamus, puis vers le cortex, où la douleur devient consciente.
La corne dorsale n'est pas un simple relais passif. C'est un véritable centre de traitement, capable d'amplifier ou d'atténuer le signal avant qu'il n'atteigne le cerveau. Dès 1965, Ronald Melzack et Patrick Wall proposaient la « théorie du portillon » (gate control theory) : les signaux tactiles transmis par les grosses fibres Aβ peuvent « fermer la porte » aux signaux douloureux des fibres C. C'est pourquoi frotter une zone douloureuse procure un soulagement transitoire.
Ce portillon est le premier niveau de modulation. Mais il en existe un second, plus puissant, et c'est celui qui dysfonctionne dans la douleur chronique.
La modulation descendante : le frein naturel de la douleur
Le cerveau ne se contente pas de recevoir passivement les signaux de douleur. Il envoie des commandes descendantes vers la moelle épinière pour les moduler, en les amplifiant ou en les supprimant selon le contexte. C'est le système de modulation descendante, et son dysfonctionnement est au cœur de la douleur chronique [1].
Le circuit principal implique deux structures :
① La substance grise périaqueducale (PAG), située dans le mésencéphale, intègre les signaux émotionnels, cognitifs et sensoriels. Elle reçoit des afférences du cortex préfrontal, de l'amygdale et de l'hypothalamus, ce qui explique pourquoi le stress, l'attention et les émotions influencent l'intensité perçue de la douleur.
② Le bulbe rostral ventromédian (RVM), qui contient deux populations de neurones fonctionnellement opposées : les cellules ON et les cellules OFF. Les cellules OFF inhibent la transmission douloureuse dans la corne dorsale (elles « freinent » la douleur). Les cellules ON la facilitent (elles « accélèrent » la douleur). L'équilibre entre ces deux populations détermine si le signal douloureux sera atténué ou amplifié [1] [2].
En situation normale, le système est en équilibre : une douleur aiguë déclenche un freinage descendant qui limite l'emballement. Mais dans certaines conditions chroniques, cet équilibre bascule : les cellules ON deviennent dominantes, les cellules OFF s'hypoactivent, et la moelle épinière commence à amplifier des signaux qui devraient être atténués. Ce déséquilibre est modulé par de multiples neurotransmetteurs dans le circuit PAG→RVM, incluant les endocannabinoïdes qui exercent un tonus inhibiteur tonique sur les cellules ON [3].
Un soldat blessé au combat peut ne ressentir aucune douleur pendant des heures, parce que la PAG active massivement les cellules OFF via la libération d'opioïdes endogènes (endorphines). À l'inverse, une personne anxieuse en salle d'attente de dentiste perçoit une douleur amplifiée, parce que le cortex préfrontal et l'amygdale activent les cellules ON. La modulation descendante explique ces différences.
Quand le système déraille : sensitisation centrale et douleur nociplastique
Lorsqu'une douleur persiste (inflammation chronique, lésion nerveuse, état post-infectieux), les neurones de la corne dorsale subissent des modifications durables. C'est la sensitisation centrale : les neurones spinaux deviennent hyperexcitables, leur seuil d'activation s'abaisse, et des stimuli normalement non douloureux (un effleurement, un vêtement) déclenchent une douleur véritable (allodynie) [4].
Le mécanisme clé est le wind-up : une stimulation répétée des fibres C provoque une augmentation progressive de la réponse du neurone spinal, bien au-delà de ce que la stimulation périphérique justifie. Ce phénomène implique les récepteurs NMDA du glutamate et une entrée excessive de calcium dans le neurone postsynaptique, un calcium qui active des cascades enzymatiques modifiant durablement l'excitabilité neuronale [5].
En 2021, une revue publiée dans The Lancet a formalisé le concept de douleur nociplastique, un troisième type de douleur, distinct de la douleur nociceptive (lésion tissulaire) et de la douleur neuropathique (lésion nerveuse identifiable). La douleur nociplastique résulte d'un traitement central altéré de la nociception sans lésion détectable par les examens standards [4].
La fibromyalgie, certaines céphalées de tension, le syndrome du côlon irritable, la douleur pelvienne chronique et probablement une partie des douleurs post-Covid en sont des exemples. Dans ces conditions, les examens d'imagerie et de biologie reviennent normaux, mais l'IRM fonctionnelle montre une hyperactivation des circuits de la douleur et un déficit de modulation descendante inhibitrice.
C'est cette distinction (nociceptive, neuropathique, nociplastique) qui explique pourquoi les antalgiques classiques (paracétamol, AINS, opioïdes faibles) sont souvent inefficaces dans la fibromyalgie ou le Covid long : ils ciblent la périphérie ou les voies opioïdergiques, pas le dysfonctionnement central. Les gabapentinoïdes, eux, agissent directement au niveau de la corne dorsale, là où la sensitisation s'installe.
Vous vivez avec une douleur chronique ? L'app Boussole vous aide à suivre vos ressentis au quotidien, identifier vos déclencheurs et préparer vos consultations avec des données concrètes.
Découvrir l'app gratuite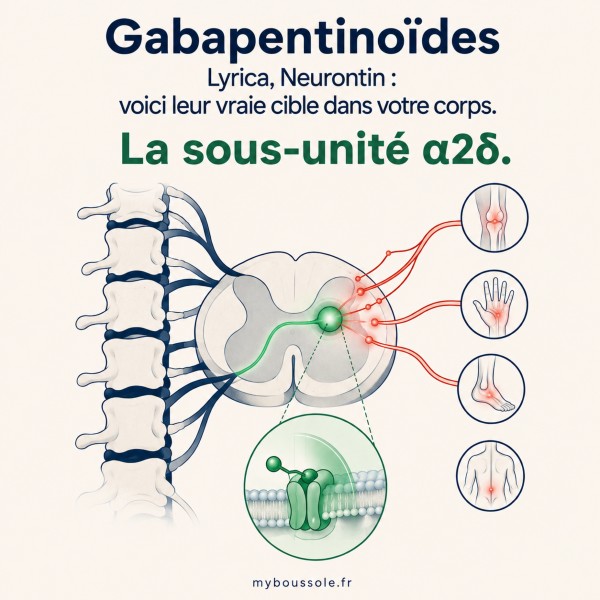
La sous-unité α2δ : la cible moléculaire des gabapentinoïdes
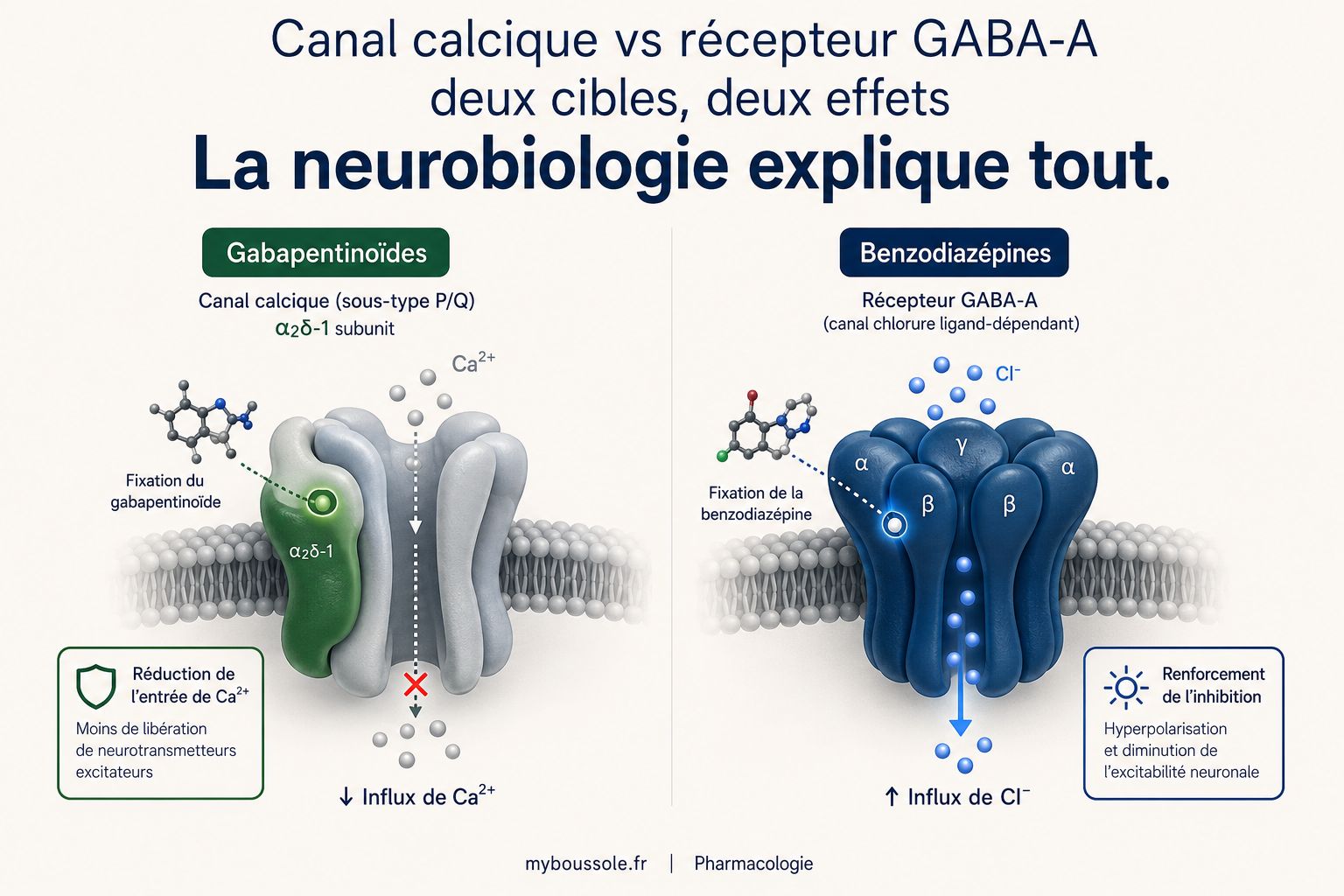
Les gabapentinoïdes ne sont ni des antalgiques classiques, ni des anti-inflammatoires, ni des opioïdes. Leur mécanisme d'action repose sur une cible moléculaire unique : la sous-unité α2δ-1 des canaux calciques voltage-dépendants (VGCC), principalement exprimée dans la corne dorsale de la moelle épinière et dans certaines régions cérébrales (cortex, amygdale, hippocampe) [6] [7].
Les canaux calciques voltage-dépendants régulent l'entrée de calcium dans les terminaisons nerveuses présynaptiques. Ce calcium est le signal qui déclenche la libération de neurotransmetteurs dans la fente synaptique. Plus il y a de calcium qui entre, plus la synapse est « bruyante », plus elle transmet de signaux excitateurs.
La sous-unité α2δ est une protéine auxiliaire de ces canaux. Elle n'est pas le canal lui-même, mais elle régule son adressage à la membrane et sa densité à la surface synaptique. En se liant à cette sous-unité, les gabapentinoïdes réduisent le trafic des canaux calciques vers la membrane présynaptique, ce qui diminue l'entrée de calcium et, en conséquence, la libération de glutamate, de substance P et de noradrénaline dans la corne dorsale [6].
La preuve formelle que cette cible est nécessaire et suffisante pour l'effet analgésique vient d'une étude publiée dans PNAS en 2006 : l'équipe de Field a créé une souris portant une mutation ponctuelle de la sous-unité α2δ-1 (R217A) qui empêche la liaison de la prégabaline, sans altérer la fonction du canal calcique lui-même. Résultat : chez ces souris mutantes, la prégabaline perd totalement son effet analgésique, anxiolytique et anticonvulsivant. La liaison à α2δ-1 est la condition sine qua non de l'activité pharmacologique [8].
Contrairement à une idée reçue, la prégabaline et la gabapentine n'agissent pas sur les récepteurs GABA. Malgré le « gaba » dans leur nom, ces molécules n'ont aucune affinité pour les récepteurs GABA-A ou GABA-B, ne modifient pas le métabolisme du GABA et n'inhibent pas sa recapture. Le nom vient de leur structure chimique (analogues structuraux du GABA), pas de leur mécanisme d'action [7] [9].
Prégabaline et gabapentine : deux profils pharmacologiques distincts
Bien que ciblant la même sous-unité α2δ, la prégabaline et la gabapentine diffèrent sensiblement sur le plan pharmacocinétique, et ces différences ont des conséquences cliniques directes [9] [6].
Biodisponibilité et absorption
La prégabaline a une biodisponibilité orale ≥ 90 %, indépendante de la dose. Sa pharmacocinétique est linéaire : doubler la dose double l'exposition plasmatique, ce qui rend la titration prévisible.
La gabapentine utilise un transporteur intestinal saturable (LAT1, le transporteur des acides aminés à chaîne latérale large). Sa biodisponibilité chute de ~60 % à 300 mg à ~33 % à 1600 mg. L'absorption est donc non linéaire, et les hautes doses ne produisent pas l'augmentation d'effet attendue, ce qui a conduit au développement de la gabapentine enacarbil (prodrogue à absorption améliorée, non disponible en France).
Puissance de liaison
La prégabaline a une affinité pour la sous-unité α2δ environ 6 fois supérieure à celle de la gabapentine. Cela se traduit par des doses efficaces plus basses (150-600 mg/j vs 900-3600 mg/j) et un délai d'action potentiellement plus court [6].
Demi-vie et schéma posologique
Les deux molécules ont des demi-vies comparables (5-7 heures). La prégabaline est administrée en 2 prises par jour, la gabapentine en 3 prises, une contrainte d'observance non négligeable pour des patients souvent polymédicamentés.
Métabolisme
Les deux sont éliminées par voie rénale sous forme inchangée : pas de métabolisme hépatique via les CYP450, pas d'interaction médicamenteuse pharmacocinétique significative. C'est un avantage majeur chez les patients polymédiqués.
L'absence d'interaction CYP450 est un atout clinique réel. Mais l'élimination rénale exclusive impose une adaptation posologique en cas d'insuffisance rénale (DFG < 60 mL/min). C'est un point souvent négligé chez les patients âgés ou insuffisants rénaux, où le risque de surdosage (et donc de sédation, de chutes) est majoré.
Efficacité clinique : fibromyalgie, douleur neuropathique et au-delà
Douleur neuropathique
La prégabaline a obtenu l'AMM européenne dans la douleur neuropathique périphérique et centrale de l'adulte. Les essais pivots montrent une réduction de la douleur statistiquement significative par rapport au placebo, avec un NNT (number needed to treat) de l'ordre de 4-7 selon les méta-analyses, ce qui signifie qu'il faut traiter 4 à 7 patients pour qu'un patient supplémentaire obtienne une réduction ≥ 50 % de sa douleur. La gabapentine montre des résultats comparables dans la neuropathie diabétique et la névralgie post-zostérienne.
Fibromyalgie
La prégabaline est le seul gabapentinoïde approuvé par la FDA dans la fibromyalgie (AMM non obtenue en Europe pour cette indication). Une méta-analyse en réseau de 2022 a comparé l'amitriptyline, la duloxétine et la prégabaline dans la fibromyalgie : les trois molécules montrent une efficacité modeste mais significative sur la douleur, le sommeil et la qualité de vie, sans différence cliniquement importante entre elles [10].
L'amélioration du sommeil (un effet collatéral de la réduction de l'hyperexcitabilité neuronale) est souvent le bénéfice le plus rapidement perçu par les patients fibromyalgiques, avant même l'effet sur la douleur.
Autres indications
La prégabaline est également utilisée dans le trouble anxieux généralisé (AMM européenne) et comme antiépileptique en association. La gabapentine est prescrite hors AMM dans les bouffées de chaleur résistantes, le syndrome des jambes sans repos et certaines douleurs centrales post-AVC.
L'efficacité des gabapentinoïdes dans la douleur chronique est modeste en termes absolus : la réduction moyenne de la douleur dans les essais est de l'ordre de 1 à 2 points sur une échelle de 10. L'effet est réel et statistiquement significatif, mais il ne constitue pas une solution complète. L'intégration dans une prise en charge multimodale (activité physique adaptée, gestion du stress, hygiène du sommeil) reste indispensable.
Limites et vigilances : effets indésirables et risque de mésusage
Effets indésirables fréquents
Les effets indésirables les plus courants sont dose-dépendants et liés à la réduction de l'excitabilité neuronale : somnolence (15-25 % des patients), vertiges (10-30 %), prise de poids (5-15 %), œdèmes périphériques, vision trouble et difficultés de concentration. Ces effets sont généralement maximaux en début de traitement et s'atténuent en 2-4 semaines, mais pas toujours.
La prise de poids, liée à une augmentation de l'appétit et probablement à des effets métaboliques centraux, est un motif fréquent d'arrêt du traitement, particulièrement chez les femmes fibromyalgiques. Pour une vue d'ensemble des mécanismes pharmacologiques impliqués dans les variations de poids, voir médicaments et poids : mécanismes de prise et de perte.
Risque de mésusage et de dépendance
Une revue systématique de 2017 portant sur 59 études a documenté le potentiel d'abus des gabapentinoïdes, particulièrement de la prégabaline à hautes doses (≥ 600 mg) [11]. Le profil de mésusage concerne principalement des patients avec antécédents d'addiction (opioïdes, benzodiazépines, alcool), et rarement les patients traités aux doses thérapeutiques pour une douleur chronique.
En Europe, une étude de 2021 a confirmé une augmentation des cas de mésusage, notamment en association avec des opioïdes, une combinaison qui majore le risque de dépression respiratoire [12]. La prégabaline a été classée comme substance contrôlée (Schedule V) aux États-Unis en 2005 et au Royaume-Uni en 2019 (Class C). En France, elle n'est pas classée comme stupéfiant mais fait l'objet d'un suivi renforcé de pharmacovigilance.
L'arrêt brutal de la prégabaline ou de la gabapentine après un traitement prolongé peut provoquer un syndrome de sevrage : insomnie, anxiété de rebond, nausées, sudation, diarrhée, rarement des convulsions. La décroissance doit être progressive (sur au moins 1 à 2 semaines) et supervisée par le prescripteur. Ce syndrome est documenté même à doses thérapeutiques et ne signifie pas nécessairement une « addiction ».
En pratique officinale, les signaux d'alerte d'un mésusage sont : des demandes de renouvellement anticipé, des ordonnances de plusieurs prescripteurs pour la même molécule, une association avec des opioïdes et/ou des benzodiazépines, et des doses inhabituellement élevées. La vigilance est particulièrement importante chez les patients avec antécédents d'addiction. Le pharmacien a un rôle de sentinelle dans la détection précoce de ces situations.
Établi : la sous-unité α2δ-1 est la cible nécessaire et suffisante de l'effet analgésique des gabapentinoïdes (preuve génétique directe chez la souris mutante, 2006). La modulation descendante PAG→RVM est un circuit validé de contrôle de la douleur. La sensitisation centrale est un mécanisme documenté en neurophysiologie de la douleur chronique.
Convergent : l'efficacité des gabapentinoïdes dans la douleur neuropathique et la fibromyalgie est appuyée par plusieurs méta-analyses, avec un effet modeste mais reproductible (NNT 4-7). Le concept de douleur nociplastique est accepté par l'IASP depuis 2017.
En débat : le mécanisme exact par lequel la liaison à α2δ réduit le trafic des canaux calciques fait encore l'objet de travaux (séquestration vs recyclage vs réduction de synthèse). Le rôle de la sous-unité α2δ-2 (exprimée dans le cervelet) dans les effets indésirables (vertiges, ataxie) est suspecté mais non confirmé.
Non résolu : pourquoi certains patients répondent remarquablement bien et d'autres pas du tout. Il n'existe pas de biomarqueur prédictif de la réponse aux gabapentinoïdes.
Les gabapentinoïdes ne sont pas des antalgiques « génériques », mais des modulateurs de la transmission synaptique. Ils agissent à l'endroit où la douleur chronique se centralise : la corne dorsale de la moelle épinière. Comprendre leur mécanisme permet de comprendre à la fois leur intérêt dans certaines douleurs centralisées et leurs limites dans les douleurs purement périphériques.
Leur place reste celle d'un outil parmi d'autres dans une prise en charge multimodale, jamais une solution isolée.
Un médicament se comprend par sa cible. Sa cible se comprend par le circuit qu'elle contrôle.Questions fréquentes
Comment la prégabaline agit-elle sur la douleur ?
La prégabaline se lie à la sous-unité α2δ-1 des canaux calciques voltage-dépendants, principalement dans la corne dorsale de la moelle épinière. En réduisant l'entrée de calcium dans les terminaisons nerveuses présynaptiques, elle diminue la libération de neurotransmetteurs excitateurs (glutamate, substance P) impliqués dans la transmission de la douleur.
Quelle est la différence entre prégabaline et gabapentine ?
Les deux ciblent la même sous-unité α2δ, mais la prégabaline a une biodisponibilité orale supérieure (≥ 90 % vs 33-66 %), une pharmacocinétique linéaire (effet prévisible selon la dose), et une puissance de liaison environ 6 fois supérieure. La gabapentine utilise un transporteur intestinal saturable qui limite son absorption à haute dose.
Qu'est-ce que la douleur nociplastique ?
La douleur nociplastique est un troisième type de douleur, distinct de la douleur nociceptive (lésion tissulaire) et neuropathique (lésion nerveuse). Elle résulte d'une altération du traitement central de la douleur, sans lésion détectable. La fibromyalgie, certaines céphalées de tension et le syndrome du côlon irritable en sont des exemples reconnus.
Les gabapentinoïdes créent-ils une dépendance ?
Les gabapentinoïdes peuvent induire une dépendance physique et un syndrome de sevrage à l'arrêt brutal. Le mésusage concerne surtout la prégabaline à hautes doses, souvent en association avec des opioïdes ou des benzodiazépines. L'arrêt doit toujours être progressif et supervisé par le prescripteur.
Suivre douleur, effets indésirables et traitements aide à préparer la consultation sans modifier seul une prescription. Boussole vous aide à suivre vos ressentis au quotidien.
Essayer gratuitementSources
- De Preter CC, Heinricher MM. « Rostral ventromedial medulla: a hub for pain modulation. » Trends in Neurosciences. 2024;47(5):295-308. PMID 38749825
- Zhang M et al. « SST neurons in PAG→RVM circuit mediate neuropathic pain. » Neuron. 2023;111(7):1087-1103. PMID 36641028
- Bouchet CA, Bhatt DK, Bhatt RR, Ingram SL. « Cannabinoid action in the descending pain modulatory circuit. » British Journal of Pharmacology. 2020;177(19):4355-4367. PMID 32004514
- Fitzcharles MA, Cohen SP, Clauw DJ et al. « Nociplastic pain: towards an understanding of prevalent pain conditions. » The Lancet. 2021;397(10289):2098-2110. PMID 34062144
- Trendafilova T et al. « Bhatt NCX3 and wind-up mechanisms in dorsal horn neurons. » Neuron. 2022;110(14):2271-2285. PMID 35705078
- Taylor CP. « Mechanisms of analgesia by gabapentin and pregabalin : calcium channel α2-δ [Cavα2-δ] ligands. » Pain. 2009;142(1-2):13-16. PMID 17126531
- Thorpe AJ, Offord J. « The α2-δ protein of voltage-dependent calcium channels. » Current Opinion in Investigational Drugs. 2010;11(7):761-770. PMID 20571971
- Field MJ et al. « Identification of the α2-δ-1 subunit of voltage-dependent calcium channels as a molecular target for pain mediating the analgesic actions of pregabalin. » Proceedings of the National Academy of Sciences. 2006;103(46):17537-17542. PMID 17088553
- Bialer M. « Why are antiepileptic drugs used for nonepileptic conditions? » Epilepsia. 2012;53 Suppl 7:26-33. PMID 23153207
- Alberti FF et al. « Amitriptyline, duloxetine, and pregabalin for fibromyalgia: a systematic review and network meta-analysis. » Pain. 2022;163(7):e845-e853. PMID 35347488
- Evoy KE, Morrison MD, Saklad SR. « Abuse and misuse of pregabalin and gabapentin. » Drugs. 2017;77(4):403-426. PMID 28144823
- Kuhn J et al. « Pregabalin and gabapentin abuse and misuse in European countries. » European Neuropsychopharmacology. 2021;43:1-10. PMID 33440453