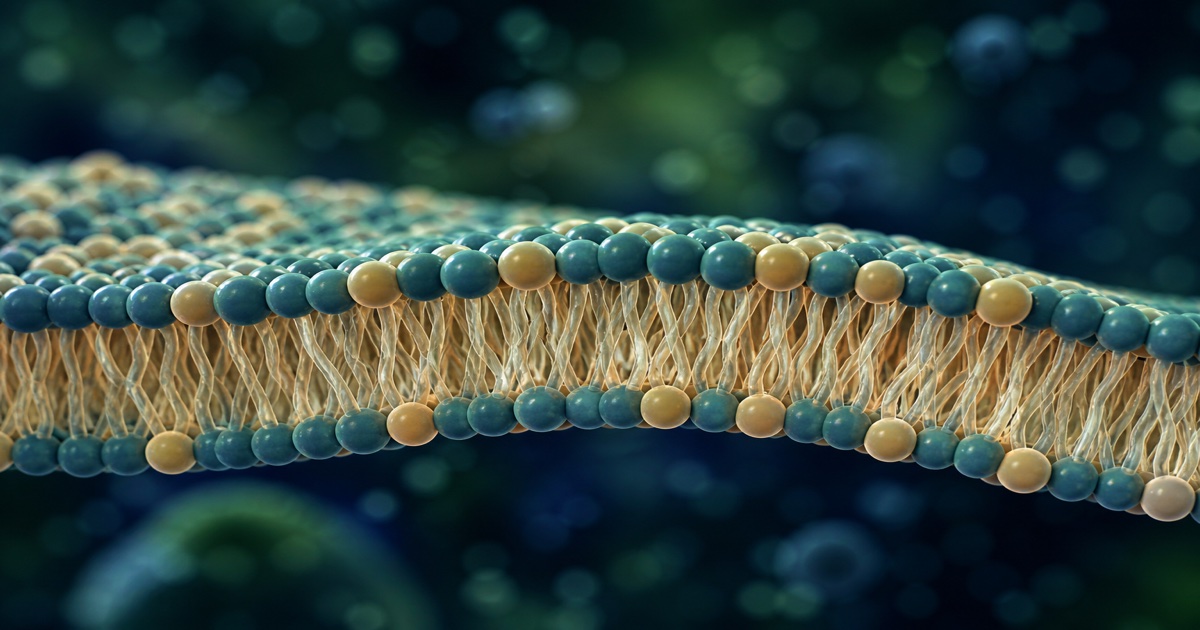
La bicouche lipidique : une architecture asymétrique au service de la signalisation
La membrane plasmique n'est pas un simple emballage. C'est une plateforme de signalisation dont la composition asymétrique entre le feuillet externe et le feuillet interne conditionne la transduction des signaux intracellulaires. C'est aussi pourquoi le bilan de cholestérol après infection ne résume qu'une petite partie de la biologie lipidique. Les quatre phospholipides majeurs sont distribués de façon non aléatoire :
- Phosphatidylcholine (PC) : feuillet externe, ~40 % des phospholipides totaux : forme structurale dominante
- Sphingomyéline : feuillet externe, ~20 % : associée aux radeaux lipidiques
- Phosphatidyléthanolamine (PE) : feuillet interne, ~25 % : précurseur de l'éthanolamine, substrat de la voie PEMT
- Phosphatidylsérine (PS) : feuillet interne, ~5-10 % : maintenu côté cytosolique par les flippases (ATP11A, ATP11C) au coût de l'hydrolyse d'ATP
- Phosphatidylinositol (PI) : feuillet interne, ~5 % : précurseur de PIP, PIP₂, PIP₃ (phosphoinositides phosphorylés)
Cette asymétrie n'est pas passive : elle coûte de l'ATP. Les flippases ATP11A et ATP11C pompent activement la PS vers le feuillet interne. Lorsque la cellule entre en apoptose, ces pompes sont inhibées et les scramblases (TMEM16F, XKR8) exposent la PS en surface : c'est le signal « eat-me » reconnu par les macrophages via les récepteurs TAM (Tyro3, Axl, Mer). Dans un contexte de fatigue cellulaire chronique ou d'infection virale persistante, cette balance flippase/scramblase est un point de vulnérabilité documenté.
La cardiolipine mérite une mention distincte : ce phospholipide à quatre chaînes acyles est quasi exclusivement mitochondrial (membrane interne). Elle stabilise les complexes de la chaîne respiratoire (I, III, IV) et le transporteur ADP/ATP (ANT). Une cardiolipine oxydée → désassemblage de la chaîne respiratoire → production de ROS amplifiée. Cependant, les auto-anticorps anti-cardiolipine ne sont pas significativement élevés dans l'EM/SFC (Nilsson 2020[13]) : contrairement aux anti-PI.
La synthèse de la PC hépatique passe par la voie PEMT (phosphatidyléthanolamine N-méthyltransférase) qui utilise trois molécules de SAMe pour méthyler la PE en PC. Ce lien entre méthylation et structure membranaire est développé dans l'article sur la SAMe : il rend compte du fait qu'un déficit en méthylation (B12, folates, SAMe) se traduit aussi par une altération de la composition membranaire.
Phosphatidylsérine et axe HPA : modulation du rétrocontrôle cortisol
La PS cérébrale joue un rôle documenté dans la modulation du rétrocontrôle de l'axe hypothalamo-hypophyso-surrénalien (HPA). Le mécanisme central implique les récepteurs aux glucocorticoïdes (GR) dont l'ancrage membranaire est PS-dépendant.
Mécanisme moléculaire
Les GR sont des récepteurs nucléaires (NR3C1), mais une fraction est associée à la membrane plasmique via un ancrage lipidique. La PS du feuillet interne fournit la surface chargée négativement nécessaire à cet ancrage. Un enrichissement en PS membranaire facilite la translocation nucléaire du GR → le rétrocontrôle négatif sur la sécrétion de CRH (hypothalamus) et d'ACTH (hypophyse) est amplifié → réduction de la sécrétion de cortisol surrénalien.
En aval, Kim 2014[6] documente que la PS cérébrale active Akt, Raf-1 et PKCδ : trois kinases impliquées dans la neuroprotection et la survie cellulaire, dont les niveaux sont perturbés dans les modèles d'hypercortisolémie chronique.
Données cliniques : RCT disponibles
Monteleone et al. 1990[1] ont administré 50 mg de PS par voie IV à 8 sujets soumis à un effort physique standardisé : l'ACTH baissait de 38 % et le cortisol de 34 % par rapport au placebo. La voie IV garantit la biodisponibilité mais ne correspond pas à un usage pratique.
Monteleone et al. 1992[2] ont répliqué avec 800 mg/j de PS soja per os pendant 10 jours chez 9 sujets entraînés : la réponse ACTH à l'exercice diminuait de 45 % et le cortisol de 39 %. Effet dose-dépendant également observé à 400 mg/j (réduction moindre).
Hellhammer et al. 2012[3] (RCT, N=40) ont utilisé une formule PS-oméga-3 en contexte de stress chronique et objectivé une réduction de la fatigue subjective en plus de la modulation hormonale. Hellhammer et al. 2014[4] (N=75, 400 mg/j PS+acide phosphatidique) ont montré un effet sur le cortisol salivaire post-stress avec une meilleure mémoire de travail à la fin de l'intervention.
La revue de Kingsley 2006[5] synthétise l'ensemble des données disponibles sur PS et HPA dans le contexte sportif : l'effet est reproductible sur le cortisol de stress aigu, moins clair sur le cortisol de repos, et nécessite au minimum 400 mg/j pendant 2 semaines pour produire un signal mesurable.
Phosphoinositides et cascade IP₃/Ca²⁺ : mécanisme cellulaire central
Le phosphatidylinositol (PI) représente environ 5 % des phospholipides de la bicouche interne, mais sa phosphorylation séquentielle génère les seconds messagers les plus importants de la signalisation calcique :
- PI → PIP (PI 4-phosphate) via PI4-kinase
- PIP → PIP₂ (PI 4,5-bisphosphate) via PIP 5-kinase : substrat direct de la PLC
- PLC (phospholipase C) clive PIP₂ → IP₃ + DAG
- IP₃ se lie au récepteur IP₃R du réticulum endoplasmique → libération de Ca²⁺ intracellulaire
- DAG active la PKC → NF-κB → transcription pro-inflammatoire
Le calcium libéré par l'IP₃R active la calmoduline (CaM) → contraction musculaire, sécrétion d'insuline, activation de la NO synthase, et modulation de l'immunité innée. Ce signal est étroitement régulé : une libération insuffisante → faiblesse musculaire et fatigue ; une libération excessive → excitotoxicité et apoptose.
Oueslati et al. 1995[9] ont montré chez des sujets soumis à un effort musculaire prolongé jusqu'à l'épuisement une réduction significative de l'inositol triphosphate (InsP₃) érythrocytaire et plaquettaire : la fatigue musculaire elle-même réduit la disponibilité de l'IP₃, créant un cercle vicieux où la contraction altère la signalisation calcique qui altère la contraction suivante.
Shen et al. 2009[8] (Nature Cell Biology) ont montré que le déficit en phosphatase MIP/MTMR14 (qui déphosphoryle PtdIns(3,5)P₂) conduit à une accumulation de PtdIns(3,5)P₂ → fuite de Ca²⁺ depuis les lysosomes → fatigue musculaire progressive dans un modèle murin. Ce mécanisme, indépendant de l'IP₃ classique, implique les endolysosomes comme réservoir calcique secondaire : un angle mécanistique sous-exploré dans la recherche EM/SFC.
Perturbations documentées dans l'EM/SFC et le Covid long
Auto-anticorps anti-phosphatidylinositol dans l'EM/SFC
Maes et al. 2007[7] ont mesuré des IgM anti-phospholipides dans une cohorte EM/SFC vs témoins sains. Les IgM anti-phosphatidylinositol (anti-PI) étaient significativement plus élevés dans le groupe EM/SFC (p < 0,001), de même que les IgM anti-phosphatidylcholine et anti-phosphatidylsérine. Les anticorps de classe IgG n'atteignaient pas la significativité, suggérant une activation préférentielle de la réponse innée-adaptative de type IgM plutôt qu'une mémoire immunologique classique.
La conséquence fonctionnelle plausible de ces anticorps anti-PI est une disruption de la cascade IP₃/Ca²⁺ : si le PI membranaire est masqué par des IgM, la PLC ne peut pas générer suffisamment d'IP₃ → libération calcique insuffisante → déficit de contraction musculaire, de sécrétion d'insuline, d'activation lymphocytaire. Ce mécanisme n'a pas encore été testé par une intervention corrective sur le PI, mais il rend compte de plusieurs manifestations caractéristiques de l'EM/SFC (faiblesse musculaire, troubles cognitifs, dysrégulation immunitaire) par un mécanisme unificateur.
PI3K/AKT/mTOR suractivé dans le Covid long
SARS-CoV-2 exploite la voie PI3K/AKT pour faciliter son entrée et sa réplication intracellulaire. Rex et al. 2021[10] ont cartographié les interactions entre les protéines virales et les voies de signalisation hôtes : PI3K/AKT apparaît comme un hub central, activé par la protéine Spike via ACE2 et par d'autres protéines virales (NSP1, NSP3, ORF3a).
Basile et al. 2021[11] ont montré que PI3K/Akt/mTOR est dérégulé de façon persistante dans les cellules immunitaires de patients Covid long : la suractivation de mTOR notamment perturbe l'autophagie et favorise un état pro-inflammatoire chronique cohérent avec la symptomatologie du Covid long.
Khezri et al. 2022[12] ont spécifiquement lié la suractivation PI3K/AKT à la coagulopathie observée dans le Covid aigu : AKT suractivé phosphoryle et inhibe la glycogène synthase kinase-3β (GSK-3β) → activation plaquettaire → thrombose microvasculaire. Ce mécanisme peut contribuer à la micro-inflammation vasculaire persistante retrouvée dans certains Covid long.
Formes, dosages et stratégie d'intégration
Phosphatidylsérine
- ① Source soja (historique) : PS de cerveau bovin a été retirée du marché (risque prion). La PS soja a fourni la majorité des données RCT (Monteleone, Hellhammer). Disponible en vrac, moins coûteuse, mais source OGM fréquente.
- ② Source tournesol (recommandée) : non-OGM, allergie soja évitée. Profil d'acides gras légèrement différent (moins de DHA que la PS soja) mais données préliminaires comparables en bioéquivalence. Forme à privilégier en pratique clinique.
- ③ Dosage efficace : 400-800 mg/j selon les études. Le seuil minimal pour un effet HPA mesurable est 300 mg/j (données d'exercice). En contexte de stress chronique ou de dysfonction cortisolique, 400 mg matin/soir semble un protocole raisonnable.
- ④ Association PS + acide phosphatidique (PA) : le protocole Hellhammer 2014 associait PS à l'acide phosphatidique (1:1 ratio) : effet synergique sur la mémoire de travail et la cognition sous stress. L'acide phosphatidique active mTORC1, ce qui potentialise le signal anabolique.
- ⑤ Précaution anticoagulants : la PS externalisée lors de l'apoptose plaquettaire fournit la surface de coagulation (prothrombinase). En théorie, une supplémentation importante pourrait interférer avec les anticoagulants. Aucun cas clinique documenté à doses thérapeutiques standards, mais vigilance chez les patients sous AVK ou NACO.
Myo-inositol : précurseur du PI membranaire
La distinction entre phosphatidylinositol (lipide membranaire incorporé dans la bicouche) et myo-inositol libre (complément disponible) est fondamentale : ce ne sont pas la même molécule. Le myo-inositol ingéré per os est phosphorylé par les cellules pour former l'inositol triphosphate libre (IP₃ cytoplsamique) ou incorporé dans le PI membranaire via la synthèse de novo, mais ce dernier processus est régulé et n'est pas une simple perfusion de PI membranaire.
- Myo-inositol 2-4 g/j : forme dominante dans les tissus (95 % de l'inositol total). Données robustes en psychiatrie (TOC, anxiété, syndrome des ovaires polykystiques). Son rôle dans la signalisation PI des pathologies chroniques est documenté in vitro mais les données d'intervention dans l'EM/SFC manquent.
- D-chiro-inositol 600 mg/j : métabolite de conversion du myo-inositol via MIOX. Rôle dans la sensibilité à l'insuline (second messager dans la voie insuline). Intéressant dans les profils avec résistance à l'insuline associée (fréquent dans l'EM/SFC).
- IP6 (hexaphosphate d'inositol) : ne pas confondre avec IP₃. L'IP6 est une réserve de phosphore, non un second messager. Données distinctes (antioxydant, chélateur de métaux).
Cardiolipine : un angle prometteur mais en attente de données dans l'EM/SFC
Les essais cliniques d'Elamipretide (SS-31), un peptide mitochondrioprotecteur qui se lie à la cardiolipine et stabilise la chaîne respiratoire, produisent des résultats positifs en insuffisance cardiaque (essai SPEED : Phase II). Dans l'EM/SFC, l'hypothèse mitochondriale implique la cardiolipine mais Nilsson 2020[13] n'a pas trouvé d'anticorps anti-cardiolipine significativement élevés (contrairement aux anti-PI). L'axe cardiolipine reste donc pertinent sur le plan mitochondrial mais ne constitue pas actuellement une piste d'intervention directe validée dans ce contexte.