Up et down-régulation des récepteurs : pourquoi ça change
Un traitement qui marchait bien et qui devient moins efficace. Un arrêt qui provoque plus de symptômes que la maladie elle-même. Ces deux situations s'expliquent souvent par le même phénomène : vos récepteurs cellulaires ajustent en permanence leur nombre et leur sensibilité au signal qu'ils reçoivent.
Vous vous demandez pourquoi un médicament semble perdre en efficacité avec le temps, pourquoi son arrêt brutal a aggravé vos symptômes, ou vous cherchez à comprendre un mécanisme cité dans d'autres articles myBoussole (ISRS, nicotine, dysautonomie) sans avoir jamais eu d'explication du principe commun qui les relie.
Ce que vous allez comprendre
Désensibilisation, down-régulation et up-régulation décrivent des changements différents, souvent confondus sous un même terme.
Pourquoi un traitement chronique perd en effet, et pourquoi son arrêt brutal peut faire plus de bruit que le traitement lui-même.
Le même principe s'applique à l'insuline ou à la leptine : ce n'est pas propre à la pharmacologie, c'est une règle cellulaire générale.
Ce mécanisme ne se mesure pas en pratique courante et ne doit pas devenir une explication universelle de tout symptôme chronique.
Glossaire rapide
- Désensibilisation : le récepteur reste présent mais devient temporairement moins réactif (phosphorylation, recrutement de β-arrestines).
- Down-régulation : le nombre total de récepteurs disponibles diminue réellement (dégradation ou réduction de leur fabrication).
- Up-régulation : le nombre de récepteurs ou leur sensibilité augmente, souvent après un blocage prolongé ou un signal trop faible.
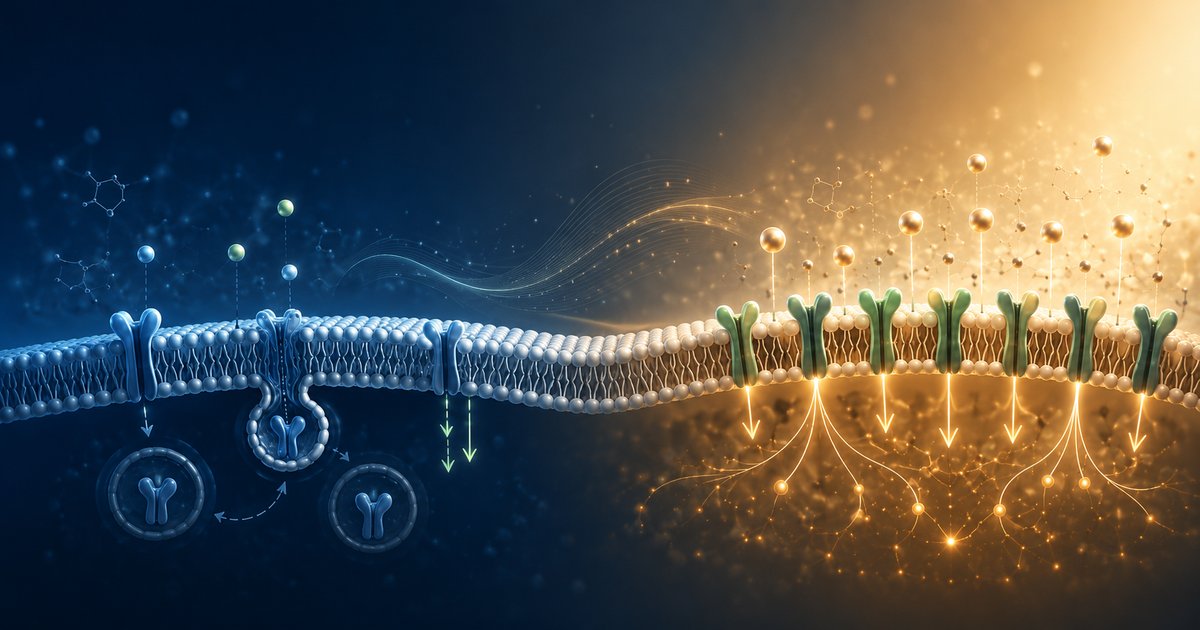
Un récepteur n'est pas un interrupteur : trois mécanismes distincts
🟢 Mécanisme établi — pharmacologie des récepteurs couplés aux protéines GUn récepteur cellulaire n'est pas un simple interrupteur : selon la façon dont il est stimulé, il peut devenir temporairement moins réactif (désensibilisation), voir son nombre diminuer (down-régulation) ou augmenter (up-régulation). Ces trois mécanismes distincts sont bien connus en pharmacologie et expliquent pourquoi un même médicament peut agir différemment selon les personnes et selon la durée du traitement.
Voir le détail ↓Une cellule ne reçoit pas passivement un signal : elle ajuste en permanence le nombre et la sensibilité de ses récepteurs, selon trois mécanismes distincts.
Un récepteur (adrénaline sur les récepteurs bêta, dopamine, histamine, acétylcholine, insuline) fonctionne comme une serrure activée par une clé. Mais contrairement à un interrupteur, la serrure elle-même change avec l'usage. En cas d'activation répétée ou prolongée, une cascade de phosphorylation (via des enzymes appelées GRK, pour G protein-coupled receptor kinases) recrute des protéines appelées β-arrestines sur le récepteur. Ce recrutement a été caractérisé en détail sur le récepteur µ-opioïde, où il découple le récepteur de sa voie de signalisation en quelques minutes, sans forcément le faire disparaître : c'est la désensibilisation[1]. Toutes les familles de GRK ne produisent pas exactement le même effet : certaines sous-familles favorisent davantage le recrutement des β-arrestines que d'autres, ce qui module l'intensité de la désensibilisation selon la molécule utilisée[5]. C'est aussi ce mécanisme qui explique la tolérance croisée observée entre différents opioïdes : une exposition préalable à l'un peut réduire l'efficacité d'un autre, administré ensuite pour la même douleur[2]. Si l'exposition se prolonge, une partie de ces récepteurs désensibilisés est ensuite internalisée puis dégradée : le nombre total de récepteurs disponibles diminue réellement, c'est la down-régulation. À l'inverse, quand un récepteur est bloqué ou insuffisamment stimulé pendant une période prolongée, la cellule peut augmenter le nombre de récepteurs ou leur efficacité de couplage : c'est l'up-régulation.
👁️ L'œil du Docteur en pharmacie
Deux personnes recevant la même dose du même médicament peuvent répondre différemment parce que leur gène du récepteur diffère. C'est documenté pour le récepteur bêta-2 adrénergique (ADRB2) : certains variants génétiques (haplotypes) expriment davantage de récepteurs en surface, et se désensibilisent plus ou moins vite sous traitement chronique par bêta-agoniste[4]. Une réponse thérapeutique différente d'un proche sous le même traitement n'est donc pas forcément un problème d'observance.
Tolérance, rebond, sevrage : ce que ça change en pratique
🟢 Mécanisme établi (tolérance) — 🟠 association documentée (sevrage ISRS)Un traitement pris longtemps peut perdre en effet (tolérance) parce que le récepteur qu'il cible s'adapte à sa présence continue. À l'arrêt brutal, cette adaptation reste active un moment, ce qui peut provoquer un rebond de symptômes parfois plus fort que le problème d'origine. C'est documenté pour les bêta-bloquants et discuté pour les antidépresseurs ISRS, où le mécanisme exact reste encore débattu.
Voir le détail ↓Pour certaines classes médicamenteuses, la tolérance en cours de traitement et le rebond à son arrêt sont deux faces du même phénomène d'adaptation réceptorielle.
Pour les récepteurs bêta-2 (utilisés par les bronchodilatateurs), une stimulation prolongée entraîne une down-régulation par un micro-ARN spécifique (let-7), qui réduit la traduction du récepteur et explique la perte d'efficacité progressive (tachyphylaxie) de certains traitements inhalés[3]. À l'inverse, l'arrêt brutal d'un bêta-bloquant chronique peut provoquer une sursensibilité du système : dans un modèle animal, la réponse à un stimulant adrénergique restait significativement augmentée jusqu'à 72 heures après l'arrêt[7]. Mais cette sursensibilité n'est pas toujours une simple augmentation du nombre de récepteurs : une étude n'a trouvé aucun changement du nombre de sites de liaison malgré une sensibilité accrue de certains tissus, suggérant un changement de couplage plutôt qu'une up-régulation classique[6]. Pour les antidépresseurs ISRS, le syndrome d'arrêt est documenté cliniquement, mais son mécanisme exact reste discuté : les auteurs d'une revue de référence reconnaissent eux-mêmes qu'expliquer ce phénomène « repose sur un certain niveau de spéculation », plusieurs systèmes de neurotransmission (sérotonine, noradrénaline, acétylcholine) étant probablement impliqués simultanément[8].
Une évolution progressive d'un ressenti autour d'un changement de traitement (efficacité qui diminue, symptômes à l'arrêt) est difficile à objectiver de mémoire. Un journal daté permet de documenter la chronologie exacte à partager avec votre médecin.
Découvrir myBoussole⚠️ Avertissement
L'arrêt brutal d'un bêta-bloquant, d'une benzodiazépine ou de certains opioïdes après un traitement chronique peut provoquer un rebond ou un syndrome de sevrage significatif, précisément à cause des adaptations décrites dans cet article. Ce n'est jamais une décision à prendre seul : un arrêt progressif encadré par le professionnel de santé qui a prescrit le traitement est la règle, quelle que soit la molécule concernée.
Au-delà des médicaments : insuline, leptine, même logique
🟢 Mécanisme établi (récepteur insuline) — 🟠 modèle physiologique (leptine)Ce principe ne concerne pas que les médicaments : il s'applique aussi aux hormones que le corps produit lui-même. Une insuline chronique trop élevée réduit le nombre de récepteurs à l'insuline disponibles, ce qui participe à l'insulinorésistance. Un mécanisme comparable existe pour la leptine, l'hormone de la satiété : en excès prolongé, le cerveau devient moins réceptif à son signal, un phénomène qui ressemble à une adaptation plutôt qu'à une simple panne.
Voir le détail ↓Ce principe d'adaptation réceptorielle ne concerne pas que les médicaments : il s'applique aussi aux hormones que votre corps produit lui-même.
Une étude combinant analyse transcriptomique humaine (plus de 450 échantillons) et modèles cellulaires a montré qu'une hyperinsulinémie chronique réduit directement l'expression du gène du récepteur à l'insuline (INSR), via un régulateur appelé SIN3A : moins de récepteurs fonctionnels en surface, ce qui contribue à l'insulinorésistance, indépendamment de tout autre facteur[9]. Le même principe est décrit pour la leptine, l'hormone de satiété : en cas d'exposition chronique excessive, la voie de signalisation JAK2-STAT3 est freinée par des inhibiteurs cellulaires (SOCS3, PTP1B), rendant le cerveau moins réceptif au signal de satiété. Une revue de référence souligne que cette résistance apparaît aussi dans des contextes physiologiques non pathologiques (hibernation, grossesse), ce qui suggère un mécanisme adaptatif plutôt qu'une simple panne[10].
Exemple hormonal : la leptine
La leptine illustre le même principe sans nécessiter un schéma autonome : signal élevé et prolongé, activation de freins intracellulaires comme SOCS3 ou PTP1B, puis signal de satiété moins bien transmis. Le point important n'est pas de mémoriser cette cascade, mais de comprendre qu'une hormone peut aussi induire une adaptation de ses propres voies de signalisation.
⚠️ Prudence d'interprétation
Les modèles physiologiques de résistance à la leptine (hamster en photopériode courte, campagnol, rate gestante) proviennent principalement de la recherche animale. Le mécanisme moléculaire (SOCS3, PTP1B) est cohérent avec ce qui est observé dans l'obésité humaine, mais la transposition directe d'un modèle saisonnier animal à une situation clinique chronique humaine reste une extrapolation, pas une démonstration directe chez l'humain dans ce contexte précis.
À retenir en pratique
Si un traitement chronique semble perdre en efficacité, ou si son arrêt provoque un rebond de symptômes, ce n'est ni un échec personnel ni une preuve que « rien ne marche » : c'est un phénomène pharmacologique décrit et attendu, à mentionner tel quel à votre médecin ou pharmacien plutôt qu'à interpréter seul.
Pourquoi le corps fait ça : l'homéostasie, pas un bug
🟢 Principe établi — physiologie de l'homéostasieCes ajustements ne visent pas à maximiser l'effet d'un signal, mais à maintenir une réponse cellulaire proportionnée et stable dans le temps.
Une stimulation en continu, qu'elle vienne d'un médicament pris quotidiennement ou d'une hormone en excès, représente pour la cellule un signal qui sort de sa plage habituelle. Désensibiliser, réduire ou augmenter le nombre de récepteurs sont autant de façons de ramener la réponse cellulaire dans une fourchette gérable, plutôt que de la laisser s'emballer ou s'éteindre. Ce principe explique pourquoi deux personnes exposées à la même molécule pendant la même durée peuvent présenter des trajectoires différentes : leur point de départ (génétique, historique d'exposition, état inflammatoire) n'était pas identique.
Fait établi. La désensibilisation (phosphorylation GRK, recrutement de β-arrestines), la down-régulation et l'up-régulation sont trois mécanismes distincts et bien caractérisés, décrits sur les récepteurs opioïdes, bêta-2 adrénergiques et à l'insuline. La tachyphylaxie aux bronchodilatateurs et la sursensibilité à l'arrêt d'un bêta-bloquant sont documentées expérimentalement.
Hypothèse étayée. Le syndrome d'arrêt des ISRS impliquerait des adaptations combinées des systèmes sérotoninergique, noradrénergique et cholinergique, mais aucun mécanisme unique n'est confirmé chez l'humain à ce jour. La résistance à la leptine comme réponse adaptative (plutôt que dysfonction pure) s'appuie principalement sur des modèles animaux physiologiques.
Spéculation à éviter. Attribuer un symptôme de fatigue chronique, de brouillard mental ou de Covid long à « une down-régulation généralisée des récepteurs » sans dosage ni mesure directe reste une extrapolation non vérifiable en pratique clinique courante : ce mécanisme n'est actuellement pas mesurable en routine chez une personne donnée.
Ce qu'il faut retenir
Un récepteur cellulaire n'est jamais un simple interrupteur : selon la durée et l'intensité du signal reçu, il se désensibilise, réduit son nombre (down-régulation) ou l'augmente (up-régulation). Ces trois mécanismes, bien caractérisés en pharmacologie, expliquent pourquoi certains traitements chroniques perdent en effet et pourquoi leur arrêt brutal peut, selon la molécule, provoquer un rebond. Le même principe s'étend au-delà des médicaments, jusqu'aux hormones que votre corps produit lui-même comme l'insuline.
Cette compréhension n'est pas une invitation à modifier vous-même un traitement en cours. C'est un repère pour mettre des mots sur une observation réelle (perte d'efficacité, rebond au sevrage) et en discuter avec le professionnel de santé qui suit votre traitement, particulièrement pour les bêta-bloquants, benzodiazépines et opioïdes où un arrêt encadré est essentiel.
Le problème n'est pas toujours la molécule. Il est parfois dans la façon dont le récepteur a appris à s'en protéger.Questions fréquentes
Pourquoi un médicament qui marchait bien perd-il de son effet avec le temps ?
Pourquoi arrêter un traitement d'un coup peut-il aggraver les symptômes ?
La down-régulation des récepteurs explique-t-elle la fatigue chronique ou le Covid long ?
Documenter vos changements de traitement
Un journal daté aide à objectiver une perte d'efficacité progressive ou des symptômes apparus après un changement de dose, pour en parler précisément à votre médecin.
Découvrir myBoussoleSources
- Zhang J et al., Proc Natl Acad Sci U S A, 1998 — PubMed PMID 9618555
- Nowoczyn M et al., Neuropharmacology, 2013 — PubMed PMID 23792280
- Wang WCH et al., Proc Natl Acad Sci U S A, 2011 — PubMed PMID 21447718
- Panebra A et al., PLoS ONE, 2010 — PubMed PMID 20686604
- Underwood O et al., Communications Biology, 2024 — PubMed PMID 39095612
- Chess-Williams RG & Broadley KJ, J Cardiovasc Pharmacol, 1984 — PubMed PMID 6206329
- Tenner TE, Life Sciences, 1983 — PubMed PMID 6685807
- Blier P & Tremblay P, J Clin Psychiatry, 2006 — PubMed PMID 16683857
- Cen HH et al., FASEB Journal, 2022 — PubMed PMID 34921686
- Tups A, J Neuroendocrinol, 2009 — PubMed PMID 19732287